Allgemeinheit
Kraniosynostose ist der Begriff, mit dem Ärzte auf eine Abnormalität des Schädels aufgrund der vorzeitigen Fusion eines oder mehrerer Schädelnähte hinweisen.
Schädelnähte sind die fibrösen Gelenke, die die Knochen des Schädelgewölbes zusammenbinden (dh die frontalen, temporalen, parietalen und okzipitalen Knochen).
Die Kraniosynostose kann ein isoliertes Phänomen (nicht-syndromale Kraniosynostose) oder das Ergebnis bestimmter krankhafter Zustände (syndromale Kraniosynostose) sein. Unter den krankhaften Zuständen, die eine vorzeitige Fusion der Schädelnähte verursachen, sind die bekanntesten: Crouzon-Syndrom und Apert-Syndrom.
Durch die vorzeitige Verschmelzung der Schädelnähte haben die Hirnstrukturen nicht genügend Raum zum Wachsen. Dies hat verschiedene Konsequenzen, darunter vor allem die Erhöhung des Hirndrucks (intrakranielle Hypertonie).
Eine schnelle und genaue Diagnose ermöglicht die Planung einer Ad-hoc- Behandlung. Letzteres ist vom chirurgischen Typ und hat das frühzeitige Trennen von Fusesi-Nähten zum Ziel.
Erinnerungen an die Anatomie des menschlichen Schädels
Ausgestattet mit Knochen und Knorpeln ist der Schädel die Skelettstruktur des Kopfes, die das Gesicht bildet und das Gehirn, das Kleinhirn, den Hirnstamm und die Sinnesorgane schützt.

Um das Studium und das Verständnis des Schädels zu vereinfachen, haben die Anatomen darüber nachgedacht, ihn in zwei Kompartimente, Neurocranium und Splancnocranium, zu unterteilen .
neurocranium
Das Neurokranium ist die obere Schädelregion, in der sich das Gehirn und einige der wichtigsten Sinnesorgane befinden. Die wichtigsten Knochen - streng flach - sind die frontalen, temporalen, parietalen und okzipitalen Knochen; diese bilden zusammen das sogenannte Schädelgewölbe .
splanchnocranium
Das Splanchocranium oder Gesichtsmassiv ist die antero-inferiore Region des Schädels, die sich aus geraden und unebenen Knochen zusammensetzt. Stellt die Skelettstruktur des Gesichts dar und enthält daher knöcherne Elemente wie Unterkiefer, Oberkiefer, Wangenknochen, Nasenknochen usw.
Was ist Kraniosynostose?
Die Kraniosynostose ist eine seltene Anomalie des Schädels, die durch eine unnatürliche Kopfform gekennzeichnet ist, die auf die vorzeitige Verschmelzung einer oder mehrerer Schädelnähte zurückzuführen ist . Schädelnähte sind die fibrösen Gelenke, die die Knochen des Schädelgewölbes zusammenbinden (dh die frontalen, temporalen, parietalen und okzipitalen Knochen).

Von der Website: //www.wkomsi.com/
WANN SOLLTE DER VERSCHLUSS VON KRANALNÄHTEN AUFTRETEN?
Unter normalen Bedingungen tritt die Fusion von Schädelnähten in der Zeit nach der Geburt auf (Anmerkung: Einige Prozesse enden sogar im Alter von 20 Jahren). Dieser lange Prozess der Fusion ermöglicht es dem Gehirn, richtig zu wachsen und sich zu entwickeln.
Wenn, wie bei der Craniosynostose, die Fusion zu früh erfolgt - also während der vorgeburtlichen, perinatalen * oder frühen Kindheit -, unterliegen die enzephalen Elemente (Gehirn, Kleinhirn und Hirnstamm) und einige Sinnesorgane (insbesondere Augen) eine Veränderung von Form und Wachstum.
Ursachen
Der pathophysiologische Prozess, der die Kraniosynostose bestimmt, ist die vorzeitige Fusion von Schädelnähten .
Dieser Prozess kann ein isoliertes Phänomen darstellen - wobei mit "isoliert" gemeint ist, dass es nicht mit einem bestimmten Krankheitszustand in Verbindung gebracht wird - oder er kann die Folge einiger bestimmter Syndrome sein, die fast immer genetischer Natur sind.
Vor diesem Hintergrund haben die Ärzte beschlossen, die Kraniosynostose in zwei Kategorien einzuteilen:
- Nicht-syndromale Craniosynostose . Der Begriff nicht-syndrom weist darauf hin, dass die Schädelanomalie nicht mit einer Pathologie oder einem anderen physischen Defekt assoziiert ist.
- Syndromische Kraniosynostose . Der Begriff Syndrom bedeutet, dass die Schädelmissbildung das Ergebnis eines bestimmten Syndroms ist, in den meisten Fällen eines genetischen Typs.
NICHT-SINDROMISCHE KRANIOSINOSTOSE
Ärzte und Forscher haben die Ursachen der nicht-syndromalen Kraniosynostose noch nicht geklärt.
Sie schlugen verschiedene Hypothesen vor - einschließlich des Einflusses von Umweltfaktoren oder hormonellen Problemen -, aber keine dieser Theorien fand Bestätigung in den experimentellen Ergebnissen.
Um den genauen Ursprung der Anomalie zu verstehen, sind diesbezüglich weitere Untersuchungen erforderlich.
CRANIOSINOSTOSI SINDROMICA
Nach neuesten medizinischen Erkenntnissen gibt es mehr als 150 verschiedene, recht seltene Krankheitsbilder, die eine Kraniosynostose auslösen können.
Unter diesen Syndromen sind die bekanntesten und häufigsten:
- Crouzon-Syndrom . Aufgrund spezifischer Mutationen in den Genen FGFR2 (Chromosom 10) und FGFR3 (Chromosom 4) betrifft diese besondere Krankheit ein Neugeborenes alle 60.000 und führt zu Anomalien ausschließlich auf Kopf- und Gesichtsebene.
- Apert-Syndrom . Es entsteht hauptsächlich aufgrund von Mutationen im FGFR2-Gen (das gleiche wie beim Crouzon-Syndrom) und betrifft ein Neugeborenes pro 100.000.
Anders als beim Crouzon-Syndrom sind die genetischen Veränderungen von FGFR2 so, dass Missbildungen nicht nur den Schädel und das Gesicht betreffen, sondern auch Hände und Füße.
- Pfeiffer-Syndrom . Es entsteht aufgrund von Mutationen im "üblichen" FGFR2-Gen und einem Gen mit ähnlichen Funktionen, das als FGFR1 (Chromosom 8) bezeichnet wird. Die Besonderheit dieser Mutationen ist, dass sie neben den Schädel- und Gesichtsdeformitäten auch Folgendes bestimmen: Syndaktylie, Brachydaktylie, Daumen und große Zehen (unverhältnismäßig zu den anderen Fingern).
Das Pfeiffer-Syndrom tritt in einem Fall pro 100.000 Neugeborenen auf.
- Saethre-Chotzen-Syndrom . Es handelt sich um eine genetisch bedingte Erkrankung, von der etwa alle 50.000 ein Neugeborenes betroffen ist. Verursacht verschiedene Fehlbildungen in Schädel, Gesicht, Händen und Füßen. Einige spezifische Mutationen des TWIST1-Gens auf Chromosom 7 sind für das Auftreten des Saethre-Chotzen-Syndroms verantwortlich.
EPIDEMIOLOGIE VON CRANIOSINOSTOSI
Nach den neuesten Statistiken scheint ein Kind etwa alle 1800-3000 an Kraniosynostose zu leiden.
In Bezug auf das am stärksten betroffene Geschlecht haben mehrere klinische Studien gezeigt, dass 3 von 4 Patienten Männer sind. Der Grund, warum Craniosynostose in der männlichen Bevölkerung weiter verbreitet ist, ist völlig unbekannt.
Craniosynostosis Risikofaktoren.
- Niedriges Geburtsgewicht
- Frühgeburt
- Fortgeschrittenes väterliches Alter
- Mütterliches Rauchen während der Schwangerschaft
Symptome und Komplikationen
Die meisten Symptome, die bei Vorhandensein einer Kraniosynostose beobachtet werden, sind auf einen Druckanstieg im Schädel zurückzuführen . In der Medizin wird der Druckanstieg im Schädel als intrakranielle Hypertonie oder intrakranielle Hypertonie bezeichnet .
Bei Vorhandensein einer Craniosynostose ist eine intrakranielle Hypertonie die Folge der Tatsache, dass das Gehirn und andere Strukturen im Schädel nicht den richtigen Raum zum Wachsen haben, sodass sie auf die knöchernen Strukturen des Kopfes drücken.
Allerdings ist es wichtig zu bedenken, dass Kraniosynostose zu einer verringerten Entwicklung der kognitiven Fähigkeiten und einem niedrigen IQ führen kann, wenn viele Schädelnähte beteiligt sind oder wenn die Erkrankung nicht rechtzeitig behandelt wird.
SYMPTOME DER ENDOKRANEN HYPERTENSION
Die möglichen Symptome einer intrakraniellen Hypertonie sind:
- Anhaltende Kopfschmerzen. In der Regel wird es morgens und abends schlimmer.
- Sehstörungen. Sie bestehen aus Doppelsehen, verschwommenem Sehen und verschwommenem Sehen.
- Erbrechen
- Reizbarkeit
- Geschwollene oder hervorstehende Augen
- Schwierigkeiten beim Verfolgen der Bewegung von Objekten
- Hörprobleme
- Atemprobleme
- Veränderungen im mentalen Status
- papilledema
Die Anzahl der Schädelnähte, die an der Entstehung einer Kraniosynostose beteiligt sind, hat einen entscheidenden Einfluss auf das Vorliegen einer intrakraniellen Hypertonie.
Zum Beispiel haben Ärzte beobachtet, dass die Beteiligung einer einzelnen Schädelnaht bei 15% der Patienten eine intrakranielle Hypertonie induziert; während die Beteiligung von mindestens zwei Nähten bei mindestens 60% der Patienten zu einem Druckanstieg im Inneren des Schädels führt.
Bei Vorliegen einer milden Form der Kraniosynostose beginnt die intrakranielle Hypertonie problematisch zu werden und verursacht die oben genannte Symptomatik in einem Alter von etwa 4 bis 8 Jahren.
ZEICHEN DER CRANIOSINOSTOSI
Unter den Anzeichen einer Kraniosynostose sind die häufigsten:- Bildung starrer Kämme entlang der Schädelnähte
- Anomalien der kranialen Fontanellen
- Kopf mit Abmessungen, die nicht zum Rest des Körpers passen
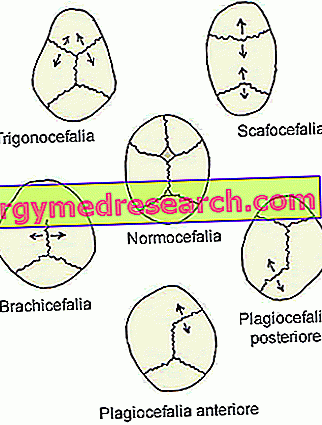
ARTEN DER KRANIOSINOSTOSE
Die Kopfform von Patienten mit Craniosynostose hängt davon ab, welche Schädelnähte sich vorzeitig geschlossen haben.
Nachdem dies beobachtet worden war, hielten es die Ärzte für angebracht, die Craniosynostose in Abhängigkeit von den beteiligten Schädelnähten in verschiedene Typen zu unterteilen.
Die Arten der Kraniosynostose sind:
- Sagittale Synostose ( Dolichozephalie oder Skaphozephalie ). Es ist die häufigste Art der Kraniosynostose; in der Tat charakterisiert es etwa die Hälfte der klinischen Fälle.
Sein Vorhandensein fällt mit dem vorzeitigen Verschluss der sagittalen Schädelnähte zusammen, die sich im oberen Teil des Schädels zwischen den Scheitelknochen befinden.
Aus //de.wikipedia.org/wiki/Plagiocephaly
- Koronale Kraniosynostose ( Brachyzephalie ) Sie ist die zweithäufigste Art der Kraniosynostose. charakterisiert etwa einen klinischen Fall alle vier.
Der Beginn besteht in der vorzeitigen Verschmelzung der koronalen Nähte, die zwischen dem Stirn- und dem Scheitelbein verlaufen.
- Metopische Synostose ( Trigonozephalie ). Es ist eine sehr seltene Art der Kraniosynostose, die nur 4-10% der Fälle unterscheidet.
Sein Aussehen fällt mit der vorzeitigen Verschmelzung der metopischen (oder frontalen) Naht zusammen, die von der Nase bis zum oberen Teil des Kopfes verläuft und den Frontalknochen in zwei Teile trennt. Im Allgemeinen verknöchert diese Naht natürlich innerhalb des sechsten Lebensjahres.
- Die lambdoide Synostose ( Plagiozephalie ). Es ist die seltenste Art der Kraniosynostose. Tatsächlich sind es nur 2-4% der klinischen Fälle.
Seine Anwesenheit beinhaltet die frühe Verschmelzung des Lambdoid-Fadens, der sich zwischen dem Scheitelbein und dem Hinterhauptbein im Hinterkopf befindet.

KOMPLIKATIONEN
Eine unbehandelte Kraniosynostose kann nicht nur die geistige Entwicklung beeinträchtigen, sondern auch Folgendes bestimmen:
- Das sogenannte obstruktive Schlafapnoe-Syndrom .
- Permanente Gesichtsveränderungen, insbesondere an Augen und Ohren.
- Permanente Missbildungen an der Schädelbasis (z. B. Missbildung oder Arnold-Chiari-Syndrom).
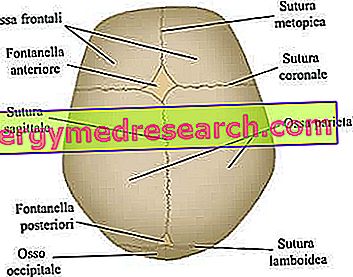
Die wichtigsten am Kraniosynostoseprozess beteiligten Schädelnähte. Von der Website: www.sciencebasedmedicine.org
- Hydrocephalus .

Diagnose
Zur Diagnose einer Kraniosynostose sind eine körperliche Untersuchung, eine Auswertung der Krankengeschichte und radiologische Bilder, die durch Röntgenstrahlen oder CT-Scans am Kopf erstellt werden, unerlässlich.
Wenn die Craniosynostose vom syndromalen Typ ist, ist es auch wichtig, den krankhaften Zustand festzustellen, der den Ausbruch verursacht hat. Daher könnten Ärzte auf Blutuntersuchungen und vor allem auf genetische Beratung zurückgreifen.
PRÜFUNGSZIEL
Die körperliche Untersuchung besteht aus einer sorgfältigen Analyse der klinischen Symptome am Kopf des Patienten durch den Arzt, bei denen der Verdacht auf eine Craniosynostose besteht.
In der Regel ist ein Kinderarzt für die Durchführung dieser wichtigen diagnostischen Untersuchung verantwortlich.
KLINISCHE GESCHICHTE
Die Auswertung der Anamnese ist für diagnostische Zwecke wichtig, da sie Fragen zu den Risikofaktoren einer Kraniosynostose enthält.
Daher wird der Arzt (in der Regel immer ein Kinderarzt) untersuchen, ob:
- Das Baby wird früh geboren oder hat ein geringes Gewicht.
- Wie alt war der Vater zum Zeitpunkt der Empfängnis?
- Wenn die Mutter während der Schwangerschaft geraucht hat.
RADIOLOGISCHE PRÜFUNGEN
Röntgenstrahlen und CT im Kopf dienen vor allem dazu, die Diagnose zu bestätigen und dem Arzt zu zeigen, welche Schädelnähte vorzeitig verschmolzen sind.
Das Wissen, um welche Schädelnähte es sich handelt, ermöglicht die Planung der am besten geeigneten chirurgischen Behandlung.
Behandlung
Die Kraniosynostose kann nur durch chirurgische Eingriffe geheilt werden .
Letzteres besteht aus einer Operation der Trennung der Schädelnähte, die früh zwischen ihnen verschmelzen.
Das endgültige therapeutische Ziel der Operation besteht darin, den enzephalen Strukturen und einigen Sinnesorganen wie den Augen den Raum zu geben, der für eine optimale Entwicklung und Funktion erforderlich ist.
BESTE ZEIT ZUM INTERVENTIONEN
Seitens der Ärzte besteht keine völlige Einigung darüber, wann die beste Zeit für eine Craniosynostose-Operation ist.
Nach Meinung einiger Experten liegt die ideale Zeit für die Operation in der späten Kindheit, wenn das Risiko eines erneuten Auftretens geringer ist (dh eine zweite vorzeitige Fusion der Schädelnähte). Im Falle eines Rückfalls muss die Operation tatsächlich wiederholt werden, und dies wird angesichts der Feinheit des Verfahrens nicht empfohlen.
Nach Ansicht anderer Experten ist die geeignetste Zeit in der frühen Kindheit (zwischen 6 und 12 Lebensmonaten), wenn der Schädel noch nicht vollständig verknöchert ist und die Knochen noch geformt werden können. Durch die Möglichkeit, Knochen zu formen (Formbarkeit), können morphologische Abnormalitäten der Knochen behoben werden, die zu ernsthaften ästhetischen Defekten und Funktionsstörungen (Kiefer oder Augen) im reiferen Alter führen können.
MÖGLICHE CHIRURGISCHE ANSÄTZE
Es gibt zwei verschiedene chirurgische Ansätze: die traditionelle Chirurgie, auch "offene" genannt, und die endoskopische Chirurgie.
- Traditionelle (oder "offene") Chirurgie .
Es sieht eine Vollnarkose (daher ist der Patient während der gesamten Operation bewusstlos) und die Durchführung eines chirurgischen Schnittes am Kopf genau dort vor, wo die radiologischen Bilder die Schädelanomalie zeigten.
Durch die Inzision am Kopf entfernt der operative Chirurg (ein Neurochirurg) den abnormalen Knochen und übergibt ihn einem Spezialisten für kraniofaziale Chirurgie, der ihn modifiziert und ihm eine Form verleiht, die die normale Entwicklung der Gehirnstrukturen ermöglicht.
Nach der Modifikation setzt der Neurochirurg den Knochen wieder in seine ursprüngliche Position ein und schließt die Inzision mit Stichen.
Wie bei vielen traditionellen Operationen ist die "offene" Operation etwas invasiv; Vorteilhaft ist jedoch, dass die Knochenstruktur präzise und mit guten Ergebnissen verändert werden kann.
- Endoskopische Eingriffe .
Dabei wird ein Endoskop verwendet, ein Werkzeug, das einem flexiblen Schlauch ähnelt, an einem Ende mit einer Glasfaserkamera ausgestattet und mit einem Monitor verbunden ist.
Aus operativer Sicht besteht es darin, das Endoskop in eine am Schädel angebrachte Öffnung einzuführen und mittels des Endoskops selbst die Fusesi-Naht frühzeitig zu trennen.
Dank der Bilder, die die Kamera auf den extern angeschlossenen Monitor projiziert, gelingt es dem Neurochirurgen, sich im Kopf zu orientieren.
Der endoskopische chirurgische Eingriff ist entschieden weniger invasiv als der "offene" Eingriff (die Hospitalisierungsdauer ist ebenfalls kürzer), hat jedoch zwei Nachteile: Er ist nur für Patienten von wenigen Monaten indiziert (6 im Allgemeinen) welche formbare Knochen besitzen; ist einem höheren Risiko eines erneuten Auftretens ausgesetzt.
POSTOPERATIVE PHASE
Im Allgemeinen muss ein Patient mit Craniosynostose, der einer Operation unterzogen wurde, nach der Operation etwa 4 bis 5 Tage im Krankenhaus bleiben. Während dieser Zeit überwachen der Neurochirurg und seine Mitarbeiter regelmäßig ihre Vitalfunktionen und prüfen, ob alles in Ordnung ist.
Nach dem Rücktritt ist eine Reihe von regelmäßigen Kontrolluntersuchungen vorgesehen, die zunächst halbjährlich und dann mit dem Wachstum des Patienten jährlich durchgeführt werden.
Prognose
Die Prognose hängt von verschiedenen Faktoren ab, darunter:
- Die Ursachen für die Craniosynostose. Einige genetisch bedingte Krankheiten, die für diese Anomalie verantwortlich sind, sind sehr ernst und haben eine schlechte Prognose.
- Die Position der Nähte verschmolz früh. Wenn sich die Nähte in Positionen befinden, die für den Neurochirurgen "unangenehm" zu erreichen sind, wird die Craniosynostose-Intervention kompliziert und liefert möglicherweise nicht die gewünschten Ergebnisse.