Allgemeinheit
Morbus Fabry ist eine seltene Erbkrankheit, die durch die Mutation des GLA-Gens verursacht wird.

Abbildung: Struktur von Alpha-Galactosidase A.
Das GLA-Gen befindet sich auf dem X-Chromosom und kodiert für ein Enzym namens Alpha-Galactosidase A. Dieses Enzym spielt eine grundlegende Rolle beim Abbau eines Lipids, das als Globotriesosylceramid bekannt ist.
Bei Menschen mit Morbus Fabry wirkt das Enzym Alpha-Galactosidase A schlecht; dementsprechend. Die Globotriesosylceramid-Moleküle neigen dazu, sich in einigen intrazellulären Organellen - den Lysosomen - abnormal anzusammeln und leiden stark unter den beteiligten Zellen.
Morbus Fabry ist verantwortlich für klinische Manifestationen auf neurologischer, dermatologischer, okularer, gastrointestinaler, zerebrovaskulärer, renaler und kardialer Ebene.
Um das Fabry-Syndrom mit absoluter Sicherheit zu diagnostizieren, ist ein geeigneter Gentest unerlässlich.
Derzeit gibt es keine Therapien, die das Fabry-Syndrom spezifisch heilen könnten, sondern nur symptomatische Behandlungen (dh zur Linderung der Symptome).
Unter diesen symptomatischen Behandlungen ist die Enzymersatztherapie am wichtigsten, bei der ein im Labor hergestelltes Analogon des Enzyms Alpha-Galactosidase A verabreicht wird.
Was ist Morbus Fabry?
Morbus Fabry ist die genetisch bedingte Erbkrankheit, die aus der Ansammlung eines bestimmten Lipidtyps, Globotriesosilceramid, in der Wand von Blutgefäßen, in Geweben und Organen resultiert.
Morbus Fabry ist eine Sphingolipidose und gehört zur heterogenen Gruppe der sogenannten lysosomalen Speicherkrankheiten .
Bedeutung von Sphingolipidose und lysosomaler Speicherkrankheit
Kurz gesagt, sind lysosomale Speicherkrankheiten eine Gruppe von etwa 50 seltenen Erbkrankheiten, die gemeinsam die Fehlfunktion einer bestimmten Kategorie intrazellulärer Organellen aufweisen: Lysosomen . Diese Fehlfunktion hängt von einem Enzymmangel ab und führt zu einer abnormalen Anreicherung von Lipiden oder Glykoproteinen in den Lysosomen selbst mit dem Ergebnis eines Verlustes der Zellfunktion.
Wenn wir uns dann der Sphingolipidose zuwenden, handelt es sich um besondere lysosomale Speicherkrankheiten, die durch die schädliche Anhäufung bestimmter Sphingolipide in den Lysosomen gekennzeichnet sind.
Ein Sphingolipid ist ein Lipid, an dessen Molekülbildung ein Sphingosinmolekül beteiligt ist.
Was ist die Aufgabe von Lysosomen?
Lysosomen sind kleine Vesikel, die typischerweise in eukaryotischen Zellen vorhanden sind und die Aufgabe haben, fremde Moleküle, Makromoleküle oder Substanzen, die für die Zelle nicht mehr nützlich sind, abzubauen und zu verdauen.
Lysosomen können als das Verdauungssystem der eukaryotischen Zelle definiert werden.
Andere Namen von Morbus Fabry
Morbus Fabry ist unter mehreren anderen Namen bekannt:
- Anderson-Fabry-Krankheit ;
- Diffuses Angiokeratom ;
- Alpha-Galactosidase A-Mangel .
Ursachen
Voraussetzung: Das humane Chromosomenkit enthält zwei Geschlechtschromosomen: das X-Chromosom und das Y-Chromosom .
Zwei bestimmte Kombinationen von X und Y bestimmen das Geschlecht einer Person: die XX- Kombination, die das weibliche Geschlecht kennzeichnet, und die XY- Kombination, die das männliche Geschlecht kennzeichnet.
Fabry-Krankheit tritt aufgrund von Mutationen im GLA-Gen auf, das sich auf dem Geschlechtschromosom X befindet.
Unter normalen Bedingungen (daher in Abwesenheit von Mutationen) codiert (dh produziert) dieses Gen ein lysosomales Enzym, Alpha-Galactosidase A, das eine grundlegende Rolle beim Abbau des oben genannten Globotriesosylceramids, eines Sphingolipids, in einfachere Komponenten spielt.
Bei Menschen mit Morbus Fabry produziert das mutierte GLA-Gen jedoch weniger Alpha-Galactosidase A als notwendig, und dies beinhaltet die fortschreitende Akkumulation von intakten (dh nicht abgebauten) Globotriesosylceramidmolekülen im Inneren der Lysosomen, was zu Schäden und Leiden für führt die beteiligten Zellen.
Mit anderen Worten, während wir bei Menschen mit gesundem GLA den korrekten Abbau des Globotriesosylceramids in den Lysosomen beobachten, reicht bei Menschen mit mutiertem GLA der gleiche Abbauprozess nicht aus, um die Bedürfnisse einer Zelle zu befriedigen. er ist krank
Welche Organe und Gewebe sind am stärksten von GLA-Mutationen betroffen?
Bei Patienten mit Morbus Fabry sind die von GLA-Mutationen am stärksten betroffenen Zellen des menschlichen Körpers:
- Die Zellen, die die Wand der Blutgefäße bilden,
- Nierenzellen,
- Herzzellen e
- Die Zellen des Nervensystems (Neuronen).
Diese Informationen sind wichtig, um den Grund für die Symptomatik zu verstehen, die den Morbus Fabry kennzeichnet.
Erbschaft
Die Fabry-Krankheit ist ein Beispiel für eine X-chromosomale rezessive Erbkrankheit, genau wie die Hämophilie .
Die Besonderheit rezessiver Erbkrankheiten im Zusammenhang mit dem X-Chromosom besteht darin, dass alle X-Chromosomen jeder Zelle dasselbe mutierte Gen besitzen müssen, damit sie sich mit schweren Symptomen manifestieren können. Dies hat mehrere Auswirkungen:
- Frauen mit nur einem X-Chromosom, das die genetische Mutation aufweist, sind im Allgemeinen in perfektem Gesundheitszustand (außer in seltenen Fällen), tragen jedoch die Erbkrankheit.
Bei diesen Personen - die als "gesunde Träger" bezeichnet werden - ist das völlige oder fast völlige Fehlen einer Symptomatik auf die Tatsache zurückzuführen, dass das gesunde X-Chromosom das anomale Verhalten des mutierten X-Chromosoms ausgleicht.
- Männliche Personen, die aus der Vereinigung eines gesunden Mannes und eines gesunden Trägers hervorgegangen sind, haben ein Krankheitsrisiko von 50%. Tatsächlich erben diese Probanden das X-Chromosom von der Mutter, und dieses Chromosom kann entweder das mutierte oder das gesunde sein.
Bei Männern existiert das beschriebene chromosomale Kompensationsphänomen für gesunde Träger nicht, da das X-Chromosom nur eines ist.
- Die einzigen kranken Frauen sind Frauen mit beiden mutierten X-Chromosomen. Tatsächlich gibt es in diesen Situationen keine Chromosomenkompensation und die Situation ist vergleichbar mit der des Menschen, dessen X-Chromosom die genetische Mutation darstellt.
Es sollte spezifiziert werden, dass das Vorhandensein von zwei mutierten X-Chromosomen bei einer Frau eine echte Seltenheit ist, da es die unwahrscheinliche Vereinigung zwischen einem kranken Mann und einer gesunden Trägerin erfordert.
- In Anbetracht der vorstehenden Ausführungen zum Chromosomenausgleich und der Unwahrscheinlichkeit der Vereinigung eines kranken Mannes mit einem gesunden Träger sind rezessive Erbkrankheiten im Zusammenhang mit dem X-Chromosom bei männlichen Probanden viel leichter zu beobachten.
Morbus Fabry respektiert fast vollständig jeden der oben genannten Punkte: Die einzige Diskrepanz - wenn wir es so nennen wollen - betrifft gesunde Träger, die häufiger eine nicht zu vernachlässigende Symptomatik aufweisen als andere Erbkrankheiten derselben eingeben.
Epidemiology
Morbus Fabry ist eine der häufigsten lysosomalen Speicherkrankheiten.
Wenn aufgrund der Inzidenz in der männlichen Bevölkerung eine relativ zuverlässige Schätzung vorliegt (1 Fall für jeweils 40.000-60.000 Neugeborene), liegen aufgrund der Inzidenz in der weiblichen Bevölkerung nur sehr wenige zuverlässige Daten vor.
Nach einigen Studien, die sich auf die Verbreitung in den verschiedenen Rassen der Welt beziehen, scheint es, dass Morbus Fabry alle ethnischen Gruppen der Welt betrifft, wobei die weiße Rasse leicht bevorzugt wird.
Symptome und Komplikationen
Aus symptomatologischer Sicht tritt der Morbus Fabry in der frühen Kindheit auf, mit sehr milden Manifestationen, die manchmal selbst von Seiten eines erfahrenen Auges schwer abzufangen sind.
Im Laufe der Jahre werden die Probleme immer offensichtlicher und die Betroffenen leiden mehr. In diesem Stadium ist sogar die Identifizierung der Krankheit durch Ärzte einfacher.
Die typischen Symptome und Anzeichen von Morbus Fabry sind:
- Schmerzen in den Extremitäten der Gliedmaßen (Hände und Füße) und Magen-Darm-Schmerzen .
Auch bekannt als Akroparestesie, scheinen Schmerzen in den Extremitäten der Gliedmaßen mit einer Schädigung einiger peripherer Nerven verbunden zu sein. Gastrointestinale Schmerzen scheinen dagegen auf eine Fehlfunktion der Blutgefäße zurückzuführen zu sein, die die Organe des Gastrointestinaltrakts versorgen.
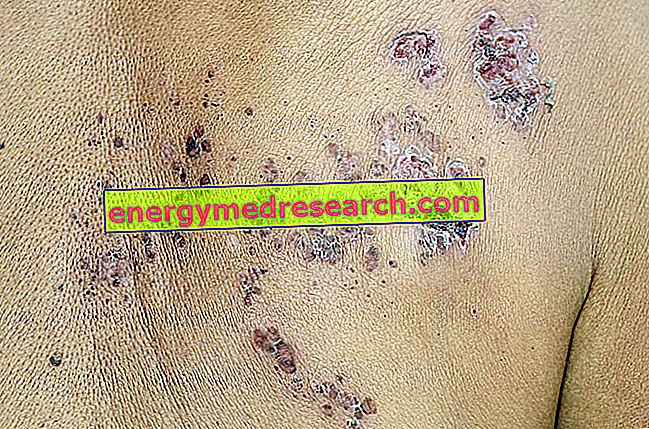
- Dunkle Flecken auf der Haut . Diese Flecken erscheinen in Clustern und werden als Angiokeratome bezeichnet .
- Anhidrose, die die Unfähigkeit ist, Schweiß abzuscheiden, oder Hypohydrose, die die abnormale Verringerung des Schweißens ist;
- Hornhauttrübung . Die Hornhauttrübung, die auch als Cornea verticillata bezeichnet wird und die Fabry-Krankheit auszeichnet, beeinträchtigt die Sehfähigkeit des Patienten nicht.
- Hörprobleme . Unter diesen sind Tinnitus und ein gewisser Grad an Taubheit;
- Schwindel ;
- Übelkeit ;
- Gefühl wiederkehrender Müdigkeit ;
- Durchfall .

Abbildung: Angiokeratom bei einem Patienten mit Fabry-Syndrom.
Komplikationen bei Morbus Fabry
In einem fortgeschrittenen Stadium neigt Morbus Fabry dazu, wichtige anatomische Organe und Strukturen wie die Nieren, das Herz und die zerebralen Blutgefäße einzubeziehen, wodurch diese geschädigt werden und ihre Funktion beeinträchtigen.
Die Hauptfolgen einer Beteiligung der Nieren, des Herzens und der zerebralen Blutgefäße durch die Fabry-Krankheit sind:
- Herzerkrankungen wie Herzinfarkt, Herzrhythmusstörungen, Herzklappenerkrankungen, restriktive Kardiomyopathie, Herzinsuffizienz usw. Sie sind das Ergebnis einer massiven Anreicherung von Globotriesosilceramid in den Herzzellen und führen häufig zum Tod.
- Nierenversagen . Ähnlich wie bei Herzerkrankungen ist es auf die massive Anreicherung von Globotriesosylceramid in Nierenzellen zurückzuführen und stellt eine wichtige Todesursache dar.
- Die Veranlagung zu Thrombose, Schlaganfall, vorübergehenden ischämischen Anfällen usw.
Komplette Sammlung aller Symptome, Anzeichen und Komplikationen von Morbus Fabry | |
| Neurologische Manifestationen |
|
Dermatologische Manifestationen |
|
Magen-Darm-Manifestationen |
|
Augenmanifestationen |
|
Zerebrovaskuläre Manifestationen |
|
Herzmanifestationen |
|
Nierenmanifestationen |
|
Diagnose
Morbus Fabry ist eine schwer zu diagnostizierende Erkrankung, insbesondere in den ersten zehn Lebensjahren des Patienten.
Im Allgemeinen sind für die Formulierung einer korrekten Diagnose des Morbus Fabry folgende Punkte von grundlegender Bedeutung: gründliche körperliche Untersuchung, sorgfältige Anamnese, Untersuchung der Familienanamnese des Patienten, enzymatischer Test zur Messung der Aktivität von Alpha-Galactosidase A in Leukozyten und ein Gentest.
Letzteres ist der diagnostische Test, der im Verdachtsfall jeden Zweifel beseitigt und es ermöglicht, die genauen Konnotationen der Mutation gegen das GLA-Gen festzustellen.
Therapie
Die Behandlung des Morbus Fabry besteht aus einer Substitutions-Enzymtherapie, bei der dem Patienten ein im Labor hergestelltes Replikat des ursprünglich fehlenden Enzyms Alpha-Galactosidase A verabreicht wird.
Daher besteht der Zweck der Enzymersatztherapie darin, Patienten etwas zur Verfügung zu stellen, das die Funktionen des normalen physiologischen Enzyms nachahmen kann.
Die Enzymersatztherapie, die für die Fabry-Krankheit untersucht wurde, erlaubt keine Behandlung der letzteren - leider bleibt die GLA-Mutation während des gesamten Lebens erhalten -, sondern ermöglicht die Linderung der gesamten Symptomatik (insbesondere der Schmerzen) mit gutem Erfolg Ergebnisse. Weitere Informationen finden Sie in den Medikamentenblättern:
- Fabrazyme
- Replagal
- Galafold
Andere Behandlungen
Es kommt sehr häufig vor, dass Ärzte zusätzlich zur Enzymersatztherapie Folgendes verschreiben:
- Die Verabreichung von Schmerzmitteln;
- Einnahme von Medikamenten gegen Magen-Darm-Probleme;
- Die Verabreichung von blutverdünnenden Medikamenten und Medikamenten zur Regulierung des Herzrhythmus;
- Eine pharmakologische Behandlung gegen Bluthochdruck;
- Dialyse.
Bei schwerer Niereninsuffizienz könnten die Grundlagen für eine Nierentransplantation vorhanden sein .
Prognose
Für Männer mit Fabry-Syndrom beträgt die durchschnittliche Lebenserwartung 58, 2 Jahre, verglichen mit 74, 7 Jahren für gesunde Männer.
Für Frauen mit Fabry-Syndrom beträgt die durchschnittliche Lebenserwartung 75, 4 Jahre, verglichen mit 80 Jahren für gesunde Frauen.
Wie aus diesen numerischen Daten hervorgeht, ist die Krankheit bei Frauen weniger schwerwiegend als bei Männern.
Bei Menschen mit Fabry-Syndrom ist eine Herzerkrankung die häufigste Todesursache.